![]()
Gene Therapy
Advancing gene therapy research for rare diseases
Essential Components of Gene Therapy
Every gene therapy is comprised of a vector, a promoter, and a transgene, and identifying the appropriate components is critical to developing viable gene therapies. Each component has a role to play:
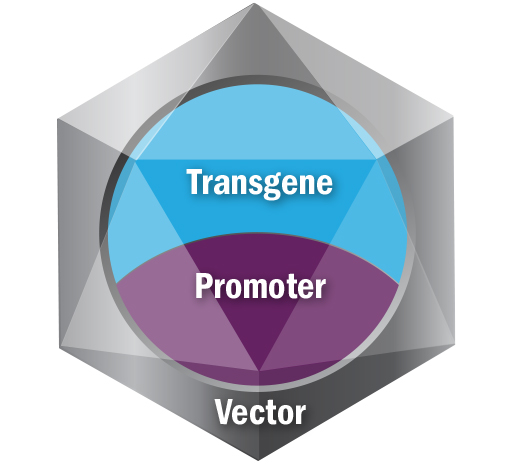
Vector: Responsible for tissue targeting and delivering the transgene and the promoter to target cells. There are several different types of vectors, including viral vectors such as adenovirus, adeno-associated virus, retrovirus or lentivirus. Other non-viral vectors used in gene therapy include bacterial, lipid- and polymer-based vectors. Patients can potentially have antibodies to some vectors and immune response is important in selecting a vector. Sarepta is currently using adeno-associated virus (AAV) vectors for our gene therapy platform.
Promoter: Responsible for selective gene expression and for driving expression in intended tissue targets.
Transgene: Responsible for producing a functioning version of the protein of interest. The transgene is genetic material introduced into targeted tissues by the vector, and its expression is driven by the promoter.
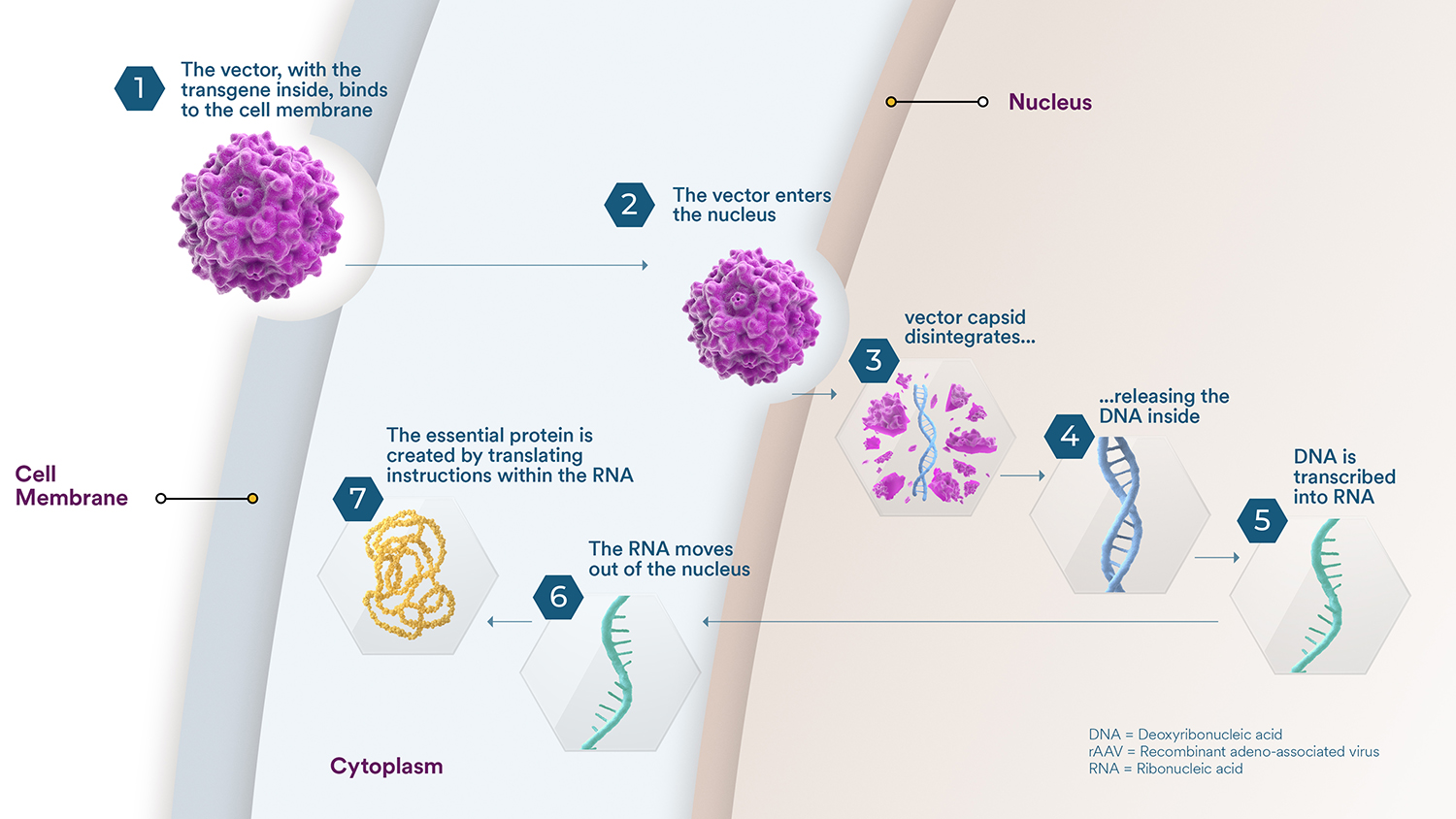

GT-FAQ: Frequently Asked Questions About Gene Therapy
As a company on an urgent mission to engineer precision genetic medicine for rare diseases that devastate lives and cut futures short, we want to help answer some frequently asked questions about gene therapy through our educational video series: GT-FAQ. These short videos, hosted by Sarepta researchers, scientists and other employees, answer some of the most common and complex questions about gene therapies in a way we can all understand.
Sarepta’s Approach to Gene Therapy for Duchenne
Sarepta Therapeutics has brought together the elements essential for successful development of gene therapy—productive collaborations, unparalleled scientific understanding, and an unwavering sense of purpose. We currently have one gene therapy on the market and additional research is underway in Duchenne.

Gene Therapy & Duchenne Brochure
Sarepta Therapeutics is leveraging our platform approach in gene therapy to research a potential treatment for Duchenne. The information in this guide provides an overview and answers some frequently asked questions about gene therapy.
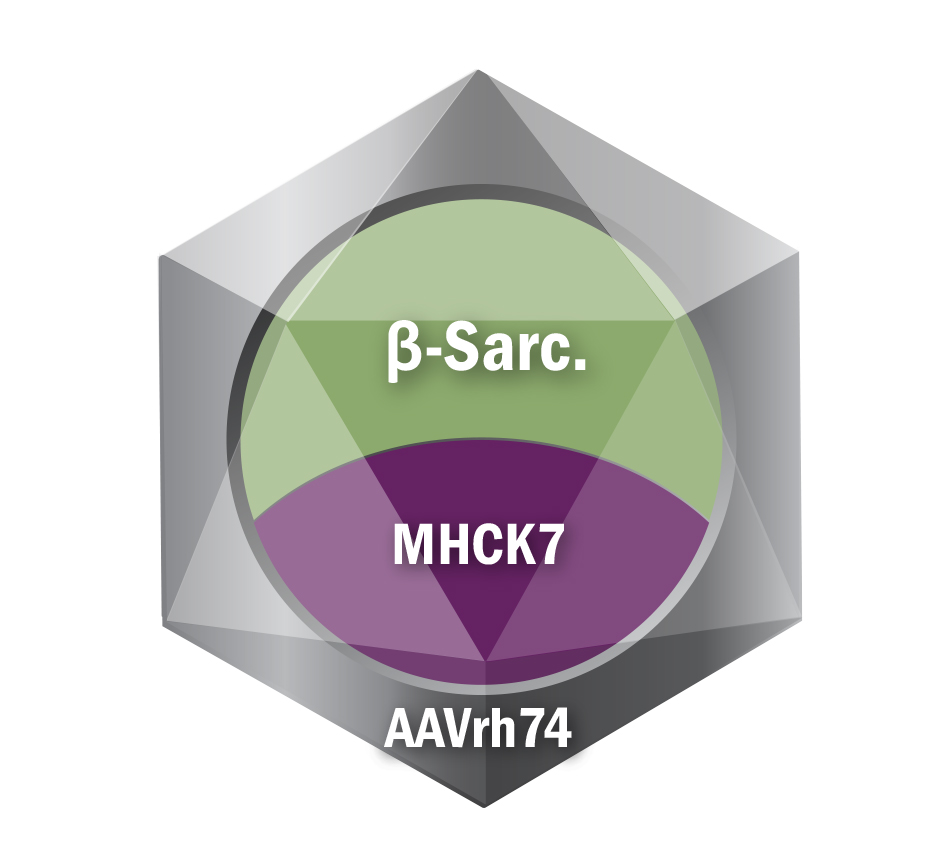
Sarepta's Gene Therapy program for LGMD type 2E
Sarepta is also pursing a therapy to treat LGMD 2E:
- LGMD type 2E beta-sarcoglycanopathy (LGMD R4)
- Vector: AAVrh74
- Promoter: MHCK7
- Transgene: beta-sarcoglycan